











请先登录您的atyun账户,方可使用该功能

审核通过后即可使用此功能,请耐心等待~






比现有技术快100万倍,哈佛大学新AI系统可预测任何蛋白质结构
2019年04月18日 由 明知不问 发表
488121
0
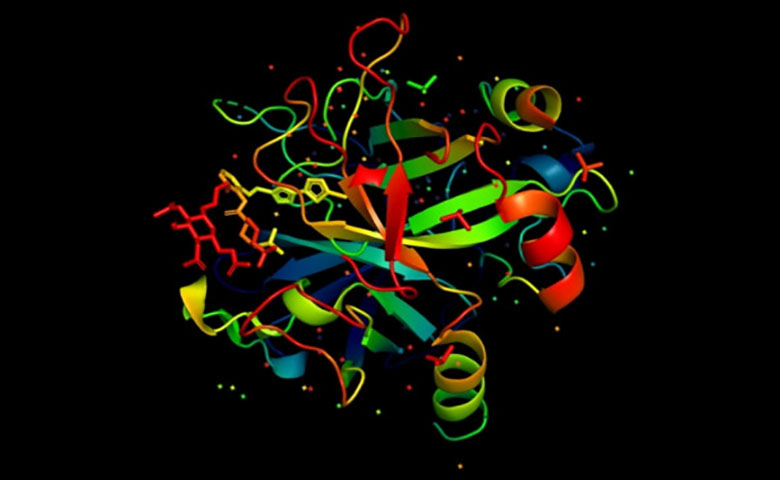 预测蛋白质的三维结构相当困难。DNA仅包含有关氨基酸残基链的信息,而不是那些链的最终形式。事实上,科学家估计,要找出一种典型蛋白质的所有可能结构需要138亿年以上的时间。
预测蛋白质的三维结构相当困难。DNA仅包含有关氨基酸残基链的信息,而不是那些链的最终形式。事实上,科学家估计,要找出一种典型蛋白质的所有可能结构需要138亿年以上的时间。如今哈佛医学院的科学家们已经用AI取得了新的进展,该系统能够有效地预测任何蛋白质的结构,并且能够在不牺牲准确性的情况下,比现有技术的系统预测快一百万倍。这项工作在Cell Systems期刊上的报告中有详细说明,软件和结果都可以通过GitHub免费获得。
HMS Blavatnik研究所系统生物学研究负责人和讲师Mohammed AlQuraishi博士表示,“在过去半个世纪中,蛋白质折叠一直是生物化学家想要解决的最重要的问题之一,新的方法代表了应对这一挑战的一种全新方式。这个问题引人注目的地方在于,它的表述相当简单:取一个氨基酸序列,然后找出它的形状。蛋白质开始作为一个非结构化的字符串必须采用3D形状,字符串可以折叠成可能的形状组合是巨大的,但我们现在可以用全新视角探索蛋白质折叠。”
AlQuraishi解释说,蛋白质由20种不同的氨基酸组成。在三维空间中,这些结构紧密地结合在一起,形成循环、螺旋、片状、扭转和其他子结构,而且它们远非随机的。氨基酸尊重物理定律,积极地寻找有利的状态,这使得它们是可预测的。
之前的方法已将新的氨基酸序列映射到预定义的模板上,或通过基因组数据筛选以识别可能一起进化的序列。例如,DeepMind的AlphaFold去年在关键性结构预测评估(CASP)蛋白质折叠竞赛中击败了98个竞争对手。
但是,正如AlQuraishi指出的那样,这些系统无法确定我们缺乏先验知识的结构,因为它们不能仅仅从序列中预测蛋白质结构。
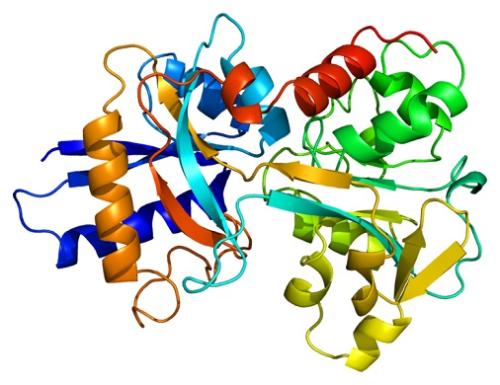
于是团队采用了一种机器学习方法,differentiable learning,其中模型通过其组件向前和向后提供数据样本来调整自身,通过这种方法发现蛋白质序列与其结构之间的关系。它们的循环几何网络仅由几千行计算机代码组成,既可以预测连接氨基酸的化学键的最可能的角度,也可以预测这些键的旋转角度。
经过几个月使用数千种蛋白质训练AI系统,系统在预测没有预先存在模板的蛋白质结构方面,超越了近年来CASP所有其他方法,此外,它还将预测结果与地面实况的蛋白质结构进行比较,以毫秒为单位检验其准确性,比现有技术快6到7个数量级,现有技术可能需要数小时。
该模型对于商业应用来说不够精准,目前,它落在6埃左右,相当于0.1纳米(解决蛋白质的完整原子结构需要大约一到两个埃)。
但AlQuraishi认为,有很多机会可以优化这种方法,例如进一步整合化学和物理规则。他说,该系统可以补充现有的计算和物理方法,以确定比以前更广泛的蛋白质结构。“我希望这种方法与已开发的所有其他优秀的方法相结合,也许在不久的将来能够准确有效地预测蛋白质折叠。”

欢迎关注ATYUN官方公众号
商务合作及内容投稿请联系邮箱:bd@atyun.com








广告

写评论取消
回复取消